Run MetalDock
MetalDock is a Python program that performs molecular docking of metal-organic compounds. It uses a configuration file ('.ini') to set various parameters for the docking procedure.
To dock organometallic compounds, follow these simple steps:
- Prepare an XYZ file of the compound you want to dock.
- Prepare a PDB file of the protein, DNA, or biomolecule you want to interact with.
- Create an input file with the desired parameters. You can find examples in the 'input_examples' directory of the GitHub repository.
How to run MetalDock docking procedure?
You can run MetalDock with a single command. Simply provide the path to your input configuration file (e.g., input.ini) as shown below:
metaldock -i input.ini -m dock
For a detailed description of the output directories and files please see the output chapter.
What basis set and functional should I use?
Choosing the correct settings for the quantum mechanical calculation can be tricky. Luckily, there have been numerous reviews on this subject that can help you with choosing the correct settings for your system. I would like to recommend the following two papers, for the less experienced quantum chemistry users:
Best-Practice DFT Protocols for Basic Molecular Computational Chemistry
Which functional should I choose?
Docking workflow
A workflow for the docking procedure is schematically given below.

How to run Monte Carlo Optimisation scheme?
This protocol describes the steps to prepare input files for Monte Carlo parameter optimization after running the MetalDock protocol for each compound in your dataset.
Prerequisites
- MetalDock installed and configured
- A dataset of compounds to be processed
- Completed MetalDock runs for each compound
Step-by-Step Instructions
1. Set Up the Run Directory & Parameter File
Create a directory where you are going to run the Monte Carlo optimization protocol in.
mc
cd mc
Copy to this folder from the /MetalDock/src/metal_dock/metal_dock.dat to the mc directory and name it metal_dock_optimize.dat.
Add at the bottom of the file the following line:
atom_par M Rii 0.010 Vol -0.00110 0.0 0.0 2 -1 -1 4 # Non H-bonding
- Replace M with the metal symbol of the new metal atom that you are going to optimize
- Replace Rii with the sum of vdW radii of two like atoms (in Angstrom)
- Replace Vol with the atomic solvation volume (in Angstrom^3)
Use this parameter file as well for the next step in creating the dataset files. If you do not do this you will receive errors that the atom type is not found.
2. Set Up the Dataset Directory
Create a dataset directory in the mc directory to store your dataset:
mkdir data_set
cd data_set
3. Prepare Each Compound
For each compound, follow these steps:
- Create a directory for the compound:
mkdir compound_1
- Copy and rename the required files from the MetalDock output:
- Ligand file
cp path/to/MetalDock/output/docking/ligand.pdbqt compound_1/compound_1.pdbqt
- Protein file
cp path/to/MetalDock/output/docking/protein.pdbqt compound_1/protein_1.pdbqt
- XYZ coordinate file
cp path/to/MetalDock/output/file_prep/ligand_c.xyz compound_1/compound_1_c.xyz
Replace
path/to/MetalDock/with the actual directory where MetalDock output files are stored.
4. Repeat for All Compounds
Repeat the above steps for each compound in your dataset, renaming directories and files accordingly (e.g., compound_2, compound_3, etc.).
5. Create input.ini file
Create a monte_carlo.ini file in the mc directory. Adjust in the monte_carlo.ini file the path and set it correctly to metal_dock_optimize.dat
[MC]
mc_steps = 100
parameter_file = metal_dock_optimize.dat
6. Run the Monte Carlo optimization
Run the following command to start the optimization.
metaldock -i monte_carlo.ini -m mc
7. Schematic overview Monte Carlo optimization
Here's an schematic overview of the Monte Carlo optimisation procedure:
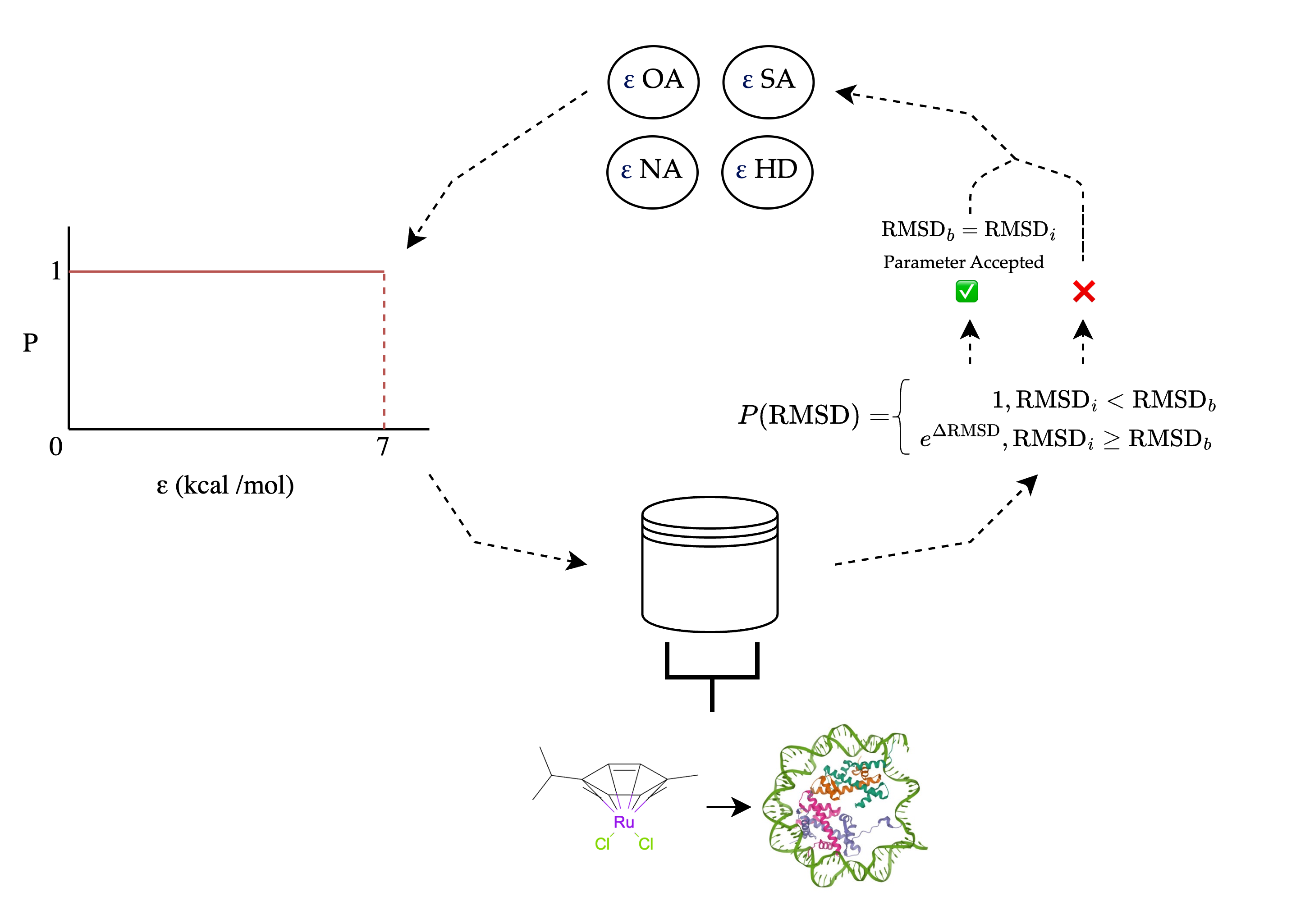
Each parameter is sequentially sampled from an equiprobable distribution. All complexes in the dataset are then docked and the average RMSD (Root Mean Square Deviation) is calculated. If the RMSD is lower than in the previous iteration, the parameter is accepted; otherwise, there is a probability of acceptance.
For more details, see the following paper.